TopMethods
Supplementary material and resources from "Identification of sparsely distributed clusters of cis regulatory elements in sets of co-expressed genes" by Gabriel Kreiman
This page adds further information to the Methods section of the main text.
1. Overall strategy
2. Nomenclature
3. Set of potentially coregulated genes
4. Sequence retrieval
5. Single motifs
6. Scanning to detect putative binding sites
7. Preliminary search for modules
8. Module enrichment
9. Figures
10. References
1. Overall strategy
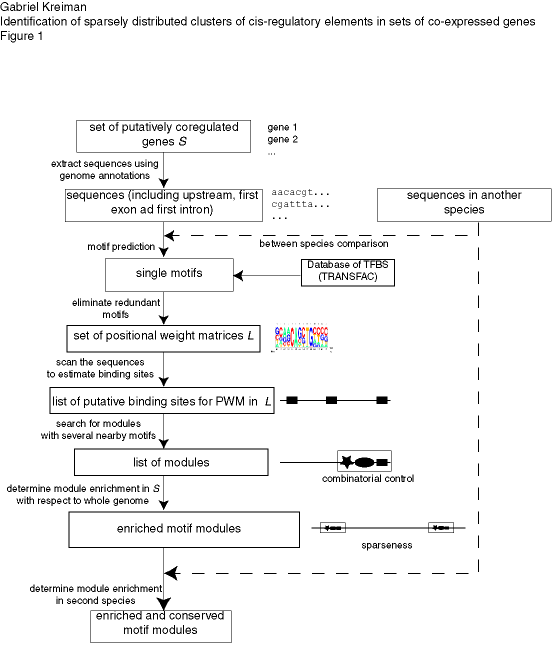
Given a set of potentially co-regulated genes, our algorithm searches for common sequence patterns that are unexpected by chance (see Figure 1 for a schematic description of the algorithm). The search is based on locating co-occurrences of putative binding sites for transcription factors within short segments of DNA. We tested the algorithm on artificial sequences with implanted modules, on random sets of genes (negative control) and in three experimentally validated systems encompassing a wide range of sequence characteristics: the CLB2 cluster in yeasts (Spellman et al., 1998), a set of genes involved in pattern formation in flies (Berman et al., 2002) and a set of genes co-expressed in the human skeletal muscle (Wasserman and Fickett, 1998). All the code is available upon request.
Figure caption: Schematic description of our approach to find cis elements in eukaryotes based on combinatorial usage of transcription factors and sparseness of the regulatory modules. The approach involves searching for co-occurrences of motifs that are highly enriched in the set of potentially co-regulated genes (S) with respect to the set of all genes in the corresponding genome. The upstream region, first exon and first intron are retrieved for each gene in S. Non-conserved sequences can be masked to reduce the level of noise. A list of individual PWMs (L) is created by (i) searching for new motifs (using the motif finding programs alignACE, MEME and MotifSampler) on the upstream, first exon and first intron sequences of the genes in S and independently (ii) from motifs from the TRANSFAC database. Modules are defined by clusters of motifs within small DNA segments and enrichment is evaluated by comparing the occurrences of the module in the set S against all the genes in the genome (see Methods). The boxes indicate the output of the previous step. The arrows indicate the processs(es) involved in each step.
3. Set of potentially coregulated genes. Positive and negative controls.
The starting point for the current algorithm is a set S of nS potentially coregulated genes. This set is assumed to be an input to the current algorithm. Several sources for S are possible including clusters of coexpressed genes from DNA microarray experiments. Here we tested the algorithm with three known sets of coregulated genes as positive controls and with random selections of mice genes from the RefSeq collection as negative controls.
Random sets
In each iteration, we randomly selected nS genes from the set of mouse RefSeq genes and applied the algorithm to search for cis regulatory elements as with the other sets. The assumption is that a random set of genes is unlikely to share a specific set of common regulatory elements (they may share general regulatory elements like the TATA box but these general elements should be present in many genes throughout the genome and therefore would not be enriched in a set of genes picked randomly). We used the following values of nS: 20, 30 and 40 genes.The process was repeated for 5 iterations (a rigorous bound on the number of modules and p values would require a much larger number of iterations, on the order of 1000 iterations. However, currently each iteration takes approximately 50 hours to run. The results for 5 iterations therefore are presented as an argument of plausibility that similar results to the ones obtained in the positive controls below are highly unlikely to arise from a random set of genes). See Performance on random sets of genes for the results.
Yeast CLB2 set
We studied the regulation of a set of genes that participate in the cell cycle in yeast. These genes show a pattern of expression that peaks in the M phase of the cell cycle (Spellman et al., 1998). The expression of several of these genes is known to be regulated by two transcription factors: MCM and SFF (Spellman et al., 1998, Koranda et al., 2000, Zhu et al., 2000). See Analysis of the CLB2 set in yeast for the results.
Drosophila pattern formation set
We studied the regulation of a set of genes involved in pattern formation in flies. These genes were taken from a previous study that combined computational and experimental work to study clustering of five known transcription factors, namely Bicoid, Caudal, Hunchback, Krüppel and Knirps (Berman et al., 2002). See Analysis of the pattern formation set in flies for the results.
Human muscle set
We studied the regulation of a set of genes expressed in the skeletal muscle in humans. Several transcription factors have been shown to play a role in the regulation of gene expression in skeletal muscle including Myf, Mef-2, SRF, Tef and Sp-1 (Wasserman and Ficket, 1998). See Analysis of the skeletal muscle set in humans for the results.
4. Sequence retrieval
Sequences were retrieved from Genbank (mm release 06/21/2003, hs release 10/22/2001), Ensembl (dm release 01-07-2003) and SGD (sc release 1/21/2003). The transcriptional start sites (TSS) as well as the boundaries for the first exon and first intron were retrieved from the corresponding genome annotations (NCBI Refseq for Mus musculus and Homo sapiens, Ensembl for Drosophila melanogaster, and SGD for Saccharomyces cerevisiae). In many cases, investigators study sequences retrieved with respect to the translation start site (which is much easier to characterize) instead of the TSS . The average annotated distance from the TSS to the ATG was 662 bp for the human muscle set and 1225 bp for the fly gene set. A comprehensive study of the distance between the TSS and the translation initial ATG in the latest release of the Drosophila genome can be found in Ohler et al., 2003. In cases of multiple alternative start sites, we used the one farthest upstream. Homologous genes in mice for the human muscle set were retrieved using the NCBI HomoloGene list. We analyzed 5000 base pairs upstream of the TSS (1000 base pairs for yeast). In addition, we also analyzed the first exon and first intron. If the length of the exon or intron was longer than 5000 base pairs, we used only the 5000 base pairs closest to the TSS. In cases of alternative splicing in the first exon/intron junction, we used the junction closest to the TSS.
5. Single motifs
In order to search for overrepresented motifs we used three available motif-finding programs: (i) alignACE (Hughes et al., 2000), (ii) MEME (Bailey et al., 1995) and (iii) MotifSampler (Thijs et al., 2002). Besides other default parameters, the parameters for alignACE were -numcols 10 and GC frequency in the corresponding genome. The parameters for MEME were -minw 6, -maxw 20, -dna, -mod tcm, -nmotifs 100, -evt 1, -minsites 3, -maxsites 500, 6th order background model, -revcomp. The parameters for MotifSampler were 6th order background model, -n 10. The background models and GC frequency were computed from the upstream sequences of all genes in the corresponding genome (using 5000 bp for each gene for mouse, human and flies and 1000 bp for yeast). The output of the motif search algorithms depends on the initial conditions. We therefore ran 10 iterations of each motif search on the same sequences. For the human muscle set, we ran the motif finding algorithms both on the raw sequences and on the sequences after masking those segments not conserved in the mouse orthologs. Sequence conservation was determined using BLAST (default parameters were -p blastn -d 0 -G 1 -E 2 -X 50 -W 11 -q -2 -r 1 -F T -e 10 -S 3). Many other approaches for sequence conservation have been proposed (see for example Wasserman et al, 2000; Loots et al, 2002; Nobrega et al, 2003). Many of these other approaches provide much more accurate models of evolutionary conservation across species for two sequences. This was not the focus of our current approach but this is a step that could be improved in future versions.
In addition, we used the available weight matrices from the TRANSFAC database of transcription factors and binding sites. We used all the entries that had the corresponding species in the species field and where a weight matrix was available from the TRANSFAC database, public release 6.0 (Wingender et al., 1997). For the muscle set, we used both the human and mouse weight matrices.
In order to remove redundancies (between PWMs obtained from different iterations of a motif-finding program, or between two different motif-finding programs or between the motif-finding programs and TRANSFAC), we used the Spearman correlation coefficient (which is more robust to outliers than the Pearson correlation coefficient). To compare two motifs, the corresponding weight matrices were converted to vectors by concatenating the rows (we refer to these vectors as "linerized weight matrices in the text").. We then computed the Spearman correlation coefficient of the two vectors and define the similarity between the two PWM as the correlation coefficient of the best alignment. Two motifs were considered to be redundant if the Spearman correlation coefficient was above 0.70 (similar results were obtained with a threshold of 0.80, not shown).
Furthermore, we only considered motifs with an information content (Stormo and Fields, 1998) larger than 0.2331 (this value corresponded to the lowest 5th percentile from the TRANSFAC database), a minimum length of 6 nucleotides and a minimum of 5 sequences used to define the weight matrix.
The resulting list L containing nL non-redundant position specific weight matrices is used to search for modules (see Preliminary search for modules below).
6. Scanning to detect putative binding sites
Detection of putative binding sites was performed by scanning the corresponding sequences and comparing to the position specific weight matrices. Click here for more details (see also Stormo and Fields, 1998). The results presented throughout the main text and in this pages correspond to those obtained using a "min_fp" threshold (i.e. the threshold that yields a low number of false negatives while accepting a larger number of false positives in the individual binding sites).
In the case of the human skeletal muscle set, we explored two possibilities for the sequences used for the scanning. In one case (masked), we masked non-conserved sequences (see Single Motifs above). In the second case (unmasked), we used the raw sequences for scanning. It is generally a matter of debate how much conservation there is for transcription regulatory mechanisms. On the one hand, important regulatory signals are expected to be conserved between humans and mice; on the other hand, we expect transcriptional regulation to be more complex in humans (at least for some cases). In the skeletal muscle set that we studied, masking yielded better results (compare masked vs unmasked for random sets of genes and masked vs. unmasked for the skeletal muscle set). However, it is unclear how well this extrapolates for other sets of genes. Therefore, the algorithm allows to turn masking on or off (see algorithm parameters).
7. Preliminary search for modules
We studied all combinations of up to 4 motifs within the list of motifs. Let nL be the total number of motifs. The total number of possible pairs is nL(nL+1)/2. Note that here we are not considering the order of the motifs to be relevant. There is still little biological data about this question and this requires further investiagion. Also, a module can be formed by instances of the same motifs, therefore allowing to consider homotypic interactions as well. The total number of possible triplets is given by nL(nL+1)(nL+2)/6. The total number of possible quadruplets is given by nL(nL+1)(nL+2)(nL+3)/24.
A potential module M was defined as a set of motifs {m1, …, mn}, with nmotifs, mi in L, that fulfilled the following requirements:
(i) Distance between adjacent motif occurrences less than maxd (we explored maxd=25, 50, 100 and 200 bp).
(ii) Maximum overlap between adjacent motifs of half the motif length.
(iii) The module had to be present in at least ntr genes in S (ntr=4 by default).
(iv) The module had to be enriched in S with respect to the background at p < 0.01 after Bonferroni correction (see definition of enrichment below).If the order constraint was not imposed the module {m1, m2, ..., mn} was considered to be equivalent to the module {m2, mn, ..., m1}, etc.
This involves a large number of computations for large values of nL. We therefore, used a preliminary definition of modules by considering a reduced background set consisting of nb genes. The background genes were randomly selected from the whole genome with the only constraint that they should not overlap the genes in S. We used nb=20 nS.
8. Module enrichment
We searched for occurrences of each of the modules selected in the previous step in the regulatory sequences (5 kb upstream + first exon + first intron) of all the genes in RefSeq for human and mice, and all annotated genes for fly and yeast (here labeled 'all genes search'). This set comprised 16979 genes in mm, 17689 genes in hs, 13639 genes in dm and 6327 genes in sc. Let Pg represent the frequency of occurrence of the module in all genes. As a null hypothesis we assumed that the module occurred with frequency Pg in S and computed the probability of obtaining the observed number of occurrences in S, here denoted x, assuming a binomial probability distribution (for large samples where Pg can be computed rather accurately, the hypergeometric distribution converges to the binomial distribution).
We reported all modules that show a probability of enrichment less than 0.01 after Bonferroni correcting by the number of putative modules. It should be noted that in the resulting list of modules, there may be a strong overlap between different modules. As an example, in the file listing the results for the fly pattern formation set with maximum distance of 100 bp and a maximum of 3 motifs, the motif number 215 appears in 3 modules from the top 10 predictions, motif number 270 appears 4 times, and even several combinatinos of motifs appear in several of the modules (e.g. the homotypic interaction of motif 270). Furthermore,even if the same motif does not appear in multiple modules, it is possible that different motifs have overlapping binding site predictions (as in the same file, when comparing the modules formed by motifs {214; 215} and the motifs {64; 215}).
When comparing multiple species, we searched for the occurrences of the module {defined by the same PSWMs) in the two species and separately computed an enrichment factor for both species.
9. Figure legends
co-occurrence positions plot
plot with all modules
plot with all interactions
10. References
Bailey, T., and Elkan, C. (1995). Unsupervised learning of multiple motifs in biopolymers using expectation maximization. Machine Learning 21, 51-80.
Berman, B. P., Nibu, Y., Pfeiffer, B. D., Tomancak, P., Celniker, S. E., Levine, M., Rubin, G. M., and Eisen, M. B. (2002). Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proceedings of the National Academy of Sciences of the United States of America 99, 757-762.
Hughes, J. D., Estep, P. W., Tavazoie, S., and Church, G. M. (2000). Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol 296, 1205-14.
Koranda, M., Schleiffer, A., Endler, L., and Ammerer, G. (2000). Forkhead-like transcription factors recruit Ndd1 to the chromatin of G2/M-specific promoters. Nature 406, 94-98.
Ohler, U., Liao, G., Niemann, H., and Rubin, G. (2003). Computational analysis of core promoters in the Drosophila genome. Genome Biology 3, 0087.
Spellman, P., Sherlock, G., Zhang, M., Iyer, V., Anders, K., Eisen, M., Brown, P., Botstein, D., and Futcher, B. (1998). Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Molecular Biology of the Cell 9, 3273-3297.
Stormo, G. D., and Fields, D. S. (1998). Specificity, free energy and information content in protein-DNA interactions. Trends Biochem Sci 23, 109-13.
Thijs, G., Marchal, K., Lescot, M., Rombauts, S., De Moor, B., Rouze, P., and Moreau, Y. (2002). A Gibbs sampling method to detect overrepresented motifs in the upstream regions of coexpressed genes. Journal of Computational Biology 9, 447-464.
Wasserman, W. W., and Fickett, J. W. (1998). Identification of regulatory regions which confer muscle-specific gene expression. J Mol Biol 278, 167-81.
Wingender, E., Kel, A., Kel, O., Karas, H., Heinemeyer, T., Dietze, P., Knuppel, R., Romaschenko, A., and Kolchanov, N. (1997). TRANSFAC, TRRD and COMPEL: Towards a federated database system on transcriptional regulation. Nucleic Acid Research 25, 265-268.
Zhu, G. F., Spellman, P. T., Volpe, T., Brown, P. O., Botstein, D., Davis, T. N., and Futcher, B. (2000). Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature 406, 90-94.
HOME